

Western blotting makes use of antibodies to determine particular person proteins from advanced samples and to carry out a semi-quantitative evaluation. First, proteins are separated from one another based mostly on their dimension by SDS-PAGE. Next, the proteins are transferred from the gel to membrane by software of {an electrical} present. Finally, the antigen-loaded blotting membrane might be detected and analyzed in accordance antigen-antibody particular binding by a particular main antibody.
Western blotting is especially used for qualitative or semi-quantitative evaluation of goal protein-specific expression, subsequent evaluation of protein-protein or protein-DNA interplay, and identification evaluation of protein modification.
Fig. 1. The transient processes of WBContents
1. Protein Sample Preparation
- Source of Protein Sample
- Proten Sample Preparation
- Selection of Protein Lysate
2. Protein Quantification
- Standard Curve Drawing with Bradford Method
- BCA Method (Please Refer to the Corresponding Kit for Instructions)
3. Protein Sample Preparation
4. SDS-PAGE
- SDS-PAGE Gel
- Electrophoresis Buffer Preparation
- Protein Marker and Loading Control
- Running the Gel
5. Transfer
- Transfer Methods
- Selection for Membranes
- Protein Separation and Membrane Transfer Condition Optimization
- Transfer Efficiency Monitoring
- Preparation of Transfer Buffer
- Transfer
6. Blocking
7. Primary Antibody Incubation
8. Secondary Antibody Incubation
- Experimental Operation
- Selection for Secondary Antibody
9. Image Development
10. Frequently Asked Questions
- High Background
- No Target Bands
- The Observed Band Size Don’t Match with the Predicted Band Size
- Other Issues
11. Protocols for ELISA, IHC, ChIP, IF, IP, FC
1. Protein Sample Preparation
1.1 Source of Protein Sample
Protein samples for western blotting may be soluble protein fluids, cell/tissue lysates or immunoprecipitated proteins. The protein loading differs from totally different samples, principally, the advisable protein loading of purified protein is not more than 100 ng, and the loading of cell/tissue lysate might be 10-40 μg.
1.2 Protein Sample Preparation
Generally, advanced protein parts are extracted from animal or plant tissues or cells, and the next ideas must be noticed throughout the extraction course of:
a. Decide the suitable extraction technique based mostly on the characters of particular person protein.
b. Use the suitable technique to maximise the extraction of goal protein.
c. Perform underneath low temperature and add protease inhibitors to stop protein degradation.
d. Choose the suitable protein lysate to keep up protein solubility.
Store Protein samples at -80℃, keep away from repeated freezing and thawing, detect as quickly as attainable.
Preparation of lysate from cell tradition
a. After the cell confluence reaches 80%, place the cell tradition dish on ice and wash the cells with ice-cold PBS for Three instances.
b. Prepare lysates which containing protease inhibitors. The generally used protease inhibitors are proven within the desk beneath (Table 2). Appropriate protease inhibitors must be chosen in line with the experimental necessities. The mostly used protease inhibitor is PMSF (working focus is 1 mM), which is extremely poisonous, so it must be self-protected when used. Its half-life in water is extraordinarily quick, so it must be added earlier than use.
c. Add 1 mL of protein lysate containing protease inhibitor to a 10 cm tradition dish, shake gently, and lyse on ice for 15-30 min.
d. Scrape adherent cells off the dish utilizing a chilly plastic cell scraper, then gently switch the cell suspension right into a 1.5 mL EP tube, then place the tube on ice. Bubbles must be averted presently.Notes:
The collected cells may also be absolutely lysed by sonication. Place the ultrasound probe in the midst of the pattern lysate, however don’t contact the tube wall or tube backside for ultrasound. The sonicator we used is Scientz JY92-IIN, with 10% energy (650 W), over 2 sec, cease for Three sec. Basically, the intracellular suspension of 1mL must be sonicated for 10-25 cycles.
e. Centrifuge at 12000 rpm for 10-15 min at 4℃.
f. Gently aspirate the supernatant to a different recent tube and place on ice for later use. Be cautious to not soak up impurities similar to lipids floating within the higher layer.
g. After protein quantification, add acceptable quantity of 6 × pattern loading buffer, and boil at 95℃ for five min, then centrifuge at 12000 rpm for 30 sec, lastly, retailer at -20℃.
Sample preparation notes:
All steps have to be operated at low temperature! Low temperature! Low temperature!
a. For the cells grown in suspension, acquire by centrifugation at 2500 rpm for Three min, adopted by cell washing and lysis procedures.
b. For drug-treated cells, particularly samples from apoptosis associated research, media supernatants also needs to be collected.
c. It shouldn’t be advisable to make use of protease to digest and acquire cells. Because it could introduce protein impurities or trigger injury to some sure proteins, particularly the membrane floor proteins, to intervene the experimental outcomes.
d. A viscous clear gel could seem within the lysate. The clear gel is a genomic DNA element. Take the supernatant for experiments. However, when the goal protein is tightly certain to the genome, the gel must be ultrasonically disrupted or syringe-sucked, then take supernatant for subsequent experiments to keep away from protein loss.
e. PMSF is unstable in aqueous answer, often it degrades by half in 30 min. The fee of lack of exercise will increase with the rise of pH worth, and the deactivation fee at 25℃ is greater than 4℃. When the pattern is processed for greater than 1 h, it must be added as soon as extra.
f. Pay consideration to the affect of cell state and the variety of cell passages. Heterogeneity exists in most cancers cells of various algebras, so the cell morphology, migration and invasion means could change, thereby make some gene expression change as nicely.
On one hand, as a result of sure heterogeneity of the cells themselves, after a interval of cultivation, the general traits of the cells are regularly modified in a means of survival of the fittest.
On the opposite hand, within the technique of cell tradition, on account of adjustments in tradition situations or the presence of exterior stimuli, similar to substitute of tradition reagents, digestion and passage, cell contamination, and a few chemical and bodily stimuli, the expression of associated genes in cells could also be affected. Ultimately have an effect on the experimental outcomes.
When utilizing tumor cells for experiments, it must be preserved first, and attempt to use related cells in the identical algebra to hold out related experimental analysis to keep away from the incidence of cell heterogeneity on account of extreme passage instances, and finally result in inconsistencies in experimental outcomes.
Preparation of lysate from tissues
a. Collect recent samples and wash with saline or PBS, then minimize into acceptable sizes. You can use a 1-2 mL homogenizer for tissue homogenate on ice, or add liquid nitrogen for grinding. It is advisable to make use of liquid nitrogen grinding, for the tissue block shouldn’t be simply broken and there’s frictional warmth era throughout the homogenization course of.
b. Prepare the lysate containing protease inhibitor.
c. Add acceptable quantity of lysate containing protease inhibitor (50 mg/500 μL) into the grinded tissue pattern, and place the tube on ice for 15-30 min for lyse, in the meantime, intermittently combine to completely lyse.Notes:
To guarantee ample lysis of tissue cells, sonication is advisable. Adjust the ultrasound system to the suitable frequency and energy (the ultrasonic energy shouldn’t be too giant, and set the ultrasonic intermittent to stop the ultrasonic probe from overheating). Place the ultrasonic probe in the midst of the pattern lysate, however don’t contact the tube wall or the underside of the tube, for ice tub ultrasound.
d. Centrifuge at 12000 rpm for 10-15 min at 4℃.
e. Remove the EP tube gently, and soak up the supernatant right into a recent tube. Be cautious to not soak up impurities similar to lipids floating within the higher layer, then place on ice for later use.
f. Centrifuge at 12000 rpm for 10-15 min at 4℃.Notes:
The tissue pattern have to be cleaned, so take away the blood vessels, and wash the blood away to keep away from interference of IgG within the pattern.
Fig. 2. The processes of lysate preparation
1.3 Selection of Protein Lysate
For most samples, RIPA lysis buffer can be utilized for speedy cell lysis.
Table 1. The parts of RIPA lysis buffer
| RIPA Lysis Buffer | |
|---|---|
| Tris-HCl | 50 mM |
| NaCl | 150 mM |
| EDTA | 1 mM |
| SDS (W/V) | 0.1% (W/V) |
| Sodium deoxycholate | 1% (W/V) |
| Triton X-100 | 1% (V/V) |
| Appropriate protease inhibitors may be added to the RIPA lysis buffer in line with the aim of experiment. |
The fundamental parts of the protein lysate and their results are as follows:
Buffer
A buffer with a sure pH vary might present a secure atmosphere for proteins and improve protein solubility as nicely. The buffers of Tris-HCl or HEPES, pH 7.Four with comparable physiological pH are generally used. The buffer of Tris-HCl (pKa = 8.1) has a pH vary of seven.0-9.2, which is delicate to temperature. The pH worth vary of HEPES (pKa = 7.55) is 6.5-8.5.
Saltion
The acceptable salt ion focus might preserve protein solubilization. The collection of 150 mM NaCl in an approximate physiological state is not going to have an effect on the disruption of proteins and protein interactions.
Chelating agent
Chelate steel ions are used to stop protein extracts from turning into too viscous, leading to decreased solubility. In addition, chelating brokers also can work together with sure enzymes to inhibit enzyme exercise.
Reducing agent
The addition of a certain quantity of decreasing agent might shield the free sulfhydryl teams on the protein from oxidation, thereby avoiding protein aggregation or denaturation. Theβ-mercaptoethanol and dithiothreitol (DTT) are generally used as decreasing brokers, the latter one is extra highly effective than the previous one. Usually, theΒ-mercaptoethanol is risky and will likely be oxidized in a short while after being added to the buffer, which might have an effect on the exercise of the protein, and its working focus is 5-20 mM/L. The DTT has a stronger decreasing means, which may type a secure intramolecular disulfide bond after oxidation with none have an effect on to the protein sulfhydryl group. Its working focus is 0.5-1 mM/L. Basically, DTT is advisable for long-term storage, however the DTT answer shouldn’t be secure and must be used proper after it was prepared.
Detergent
The detergent is a type of surfactant, and the hydrophobic section of the surfactant molecule is inserted into the phospholipid bilayer of the membrane, thereby altering its permeability and finally destroying the membrane construction. Therefore, the energy of the surfactant immediately determines the power of lysating cells. The surfactants used within the lysate may be primarily divided into two main sorts: anionic surfactants and nonionic surfactants. Commonly used surfactants are as follows:
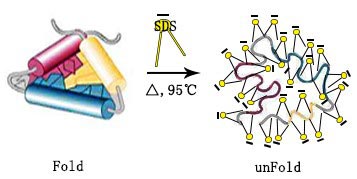
- SDS: The anionic surfactant, has a robust damaging energy, which may principally dissolve all proteins and destroy their pure conformational construction. SDS binds with protein in a ratio of 1.4:1, which may successfully cowl the cost of the protein itself. The crucial micelle temperature of SDS is a bit excessive, so precipitation might happen at low temperatures, and the precipitation will bemore obvios within the presence of potassium salts. In addition, the stronger the ionic energy of the answer, the decrease the crucial micelle focus of the ionic detergent, making the protein extra soluble.
- NaDOC: A type of ionic surfactant, which is weaker than SDS.
- Triton X-100: A type of non-ionic surfactant. It can destroy the interplay between protein and lipid, but it surely doesn’t denature the protein, or break the connection between protein and protein neither. It can protect the pure conformation of the protein. It has a decrease crucial micelle focus and two-phase separation may be noticed at 64℃.
- NP-40: A type of non-ionic surfactant, it has weak injury to the nuclear membrane, nonetheless, it has sturdy binding means to proteins, and will guarantee ample solubility and structural stability of the protein, so it’s significantly appropriate for dissolution of membrane proteins underneath non-deformation situations.
- Tween 20: A type of delicate non-ionic surfactant with poor means for protein solubilization, which doesn’t destroy protein construction and isn’t a typical element of protein lysates.
The collection of detergent relies on the character of the protein to be extracted and the aim of the experiment. There are many components must be thought of when selecting the detergent, together with absolutely lysating the cells and dissolving the protein, for the state of the extracted protein (denatured or retained in a pure state).
Protease inhibitors
A considerable amount of protease is launched when cells and tissues are destroyed throughout protein extraction course of. In order to inhibit protease exercise, the samples have to be stored at low temperature and an acceptable quantity of protease inhibitor must be added to stop degradation of the protein of curiosity.
Table 2. Commonly used protease inhibitors
| Protease inhibitor | Function | Working focus | Characters |
|---|---|---|---|
| PMSF | Serine proteases inhibitor Cysteine proteases inhibitor | 0.5-1 mM | PMSF has a brief half-life in water and must be added shortly earlier than use. Very poisonous, ought to take note of self-protection throughout experimental operation |
| APMSF | Serine proteases inhibitor | 0.4-Four mM | – |
| Pepstatin | Aspartyl proteases inhibitor | 1 μM | Store at -4℃ for 1 week, -20℃ for as much as 1 month; keep away from repeated freeze/thaw cycle. |
| Leupeptin | Serine proteases inhibitor and Cysteine proteases inhibitor | 10-100 μM | Store at -4℃ for 1 week, -20℃ for as much as 1 month; keep away from repeated freeze/thaw cycle. |
| Aprotinin | Serine proteases inhibitor | 0.01-0.03 μM | Store at -4℃ for 1 week, -20℃ for as much as 1 month; keep away from repeated freeze/thaw cycle. |
| Na3VO4 | Phosphatases inhibitor | 1 mM | Need to be activated. Add acid to regulate the pH to 10 after dissolving, and warmth to boil to colorless, cool at room temperature, then modify the pH to 10 once more. Repeat the steps till the answer stays colorless and the pH is secure at 10, aliquot and retailer at -20 ° C. |
| NaF | Phosphatases inhibitor | 10-20 mM | – |
2. Protein Quantification
In order to quantify the protein of curiosity in samples, it’s crucial to find out the quantity of whole protein in samples. The distinction within the expression stage of goal protein is mirrored when the content material of whole protein stays fixed.
Table 3 Common chemical quantification technique.
| Method | Principle | Interference components | Characters |
|---|---|---|---|
| Bradford Assay | Under acidic situations, the binding of the protein to G-250 causes a shift of most absorption wavelength of the dye. Within a sure vary, the protein content material is linear with the 595 nm absorption peak. | The assay is interfered with by sturdy alkaline buffers and excessive focus detergents | Rapid and extremely delicate, minimal detection restrict is 1μg. The protein-dye advanced has a excessive extinction coefficient and the colour is secure. The assay is especially used for the detection of primary or fragrant amino acids due to its excessive selectivity for proteins. |
| BCA assay | Cu2+ is decreased to Cu+ by protein underneath alkaline situations, the Cu+ then reacts with BCA to type a purple coloured advanced. The advanced has an absorbance at 562 nm that’s linear with protein focus. | The assay is appropriate with excessive focus detergents, and might tolerate chelating and decreasing brokers of sure concentrations. | Rapid and extremely delicate with nice anti-interference capability, restrict of detection reaches 0.5 μg. There is much less protein to protein variation in contrast with the Bradford assay |
| Lowry Assay | Under alkaline situations, Cu2+ reacts with the peptide bonds in proteins to type a posh that reduces Folin–Ciocalteu reagent, which leads to a blue color advanced. There’s a linear relationship between the shade of the color and protein focus. | The assay is appropriate for the detection of samples with excessive lipid content material and might tolerate detergent of sure concentrations. | Standard curve shouldn’t be a straight line, the shade of shade varies from protein to protein, and the detection takes longer time. |
| Ultraviolet-visible spectrophotometry(UV–Vis or UV/Vis) | Based on the bodily properties of the protein and Lambert Beer’s regulation, the absorbance at a given wavelength is linear with the protein focus. | – | The assay is interfered with by tryptophan and tyrosine ranges in several proteins |
BCA and Bradford assay are essentially the most generally used strategies for protein quantification. However, BCA assay is advisable within the presence of excessive focus detergent.
2.1 Standard Curve Drawing with Bradford Method
a. Prepare bovine serum albumin (BSA) commonplace:
Dissolve 0.05 g BSA in 5 mL PBS to acquire 10 mg/mL BSA commonplace.
b. Prepare Coomassie Brilliant Blue G-250 staining answer:
Dissolve 50 mg Coomassie Brilliant Blue G-250 in 25 mL 90% ethanol, add 50 mL phosphoric acid (85%) , and dilute with pure water to 500 mL. Keep away from mild.
c. Dilute protein samples:
Perform 1:10, 1:20, and 1:40 dilutions for protein samples.
d. Dilute BSA commonplace to the next concentrations.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Concentration(mg/mL) | 0 | 0.05 | 0.075 | 0.1 | 0.15 | 0.2 | 0.3 | 0.4 |
e. Add 20 μL every of diluted BSA commonplace answer and protein pattern to the wells of microplate strip. Then add 180 μL of G250 staining answer to every nicely and blend totally.
f. Measure absorbance with spectrophotometer at a wavelength of 595 nm and make a typical curve to calculate the protein focus.
2.2 BCA Method (Please Refer to the Corresponding Kit for Instructions)
a. Prepare the BCA working reagent by mixing BCA reagent A with reagent B in a ratio of 50:1 (V/V), and incubate for 24 h at room temperature;
b. Dissolve the usual to a ultimate focus of 0.5 mg/L with the identical solvent used for samples.
c. Pipette gradient quantity of normal answer( Zero μL, 1 μL, 2 μL, Four μL, Eight μL, 12 μL, 16 μL, 20 μL) to microplate wells, and add commonplace diluent to every nicely to ultimate quantity 20 μL;
d. Pipette 200 μL BCA working reagent to every nicely and incubate at 37℃ for 30 min;
e. Measure absorbance at 562 nm utilizing spectrophotometer;
f. Calculate protein focus based mostly on the usual curve.
3. Protein Sample Preparation
Denaturation of protein, which includes destructing protein tertiary construction and exposing antigenic epitopes, facilitates binding of antibody to the goal protein and subsequent detection. To denature protein, combine the protein pattern with 2 x pattern loading buffer at quantity ratio of 1:1 or 6 x pattern loading buffer at quantity ratio of 5:1, boil the combined answer at 95℃ for five min.
Membrane proteins are sometimes aggregated and precipitated underneath excessive temperature, they need to be handled at 37℃ for 30 min.
Table 4. The element of loading buffer
| The 6 × pattern loading buffer | |
|---|---|
| Tris-HCl (pH 6.8) | 6% (V/V) |
| SDS | 4% (W/V) |
| Bromophenol blue | 0.2% (W/V) |
| Glycerol | 20% (V/V) |
| DTT | 9% (V/V) |
| Note: loading quantity of protein pattern must be 10-40 μg per nicely. Overloading protein could cause smearing. |
4. SDS-PAGE
4.1 SDS-PAGE Gel
The polyacrylamide gel is fashioned by the polymerization of acrylamide and methylene bisacrylamide, which leads to a community of gel constructions with molecular sieving property. Proteins are encapsulated by a lot of SDS micelles with unfavorable cost, protecting the proteins’ intrinsic cost and offering the proteins with uniform charge-to-mass ratio. The decision of SDS-PAGE correlates with the focus of the cross-linker acrylamide and methylidene bisacrylamide used. The molecular sieves fashioned by totally different concentrations of cross-linking brokers have totally different pore sizes, giving a wide range of separating situations based mostly on molecular weight (particulars proven beneath).
Table 5. Gel proportion and corresponding protein dimension
| Gel proportion | Protein dimension (kDa) |
|---|---|
| 8% | 70-200 |
| 10% | 25-70 |
| 12% | 20-55 |
| 15% | 15-45 |
SDS cost impact
Table 6. Composition and performance of acrylamide gel
| Composition | Function |
|---|---|
| Acrylamide | Acrylamide monomers can polymerize to type polyacrylamide gel. |
| N,N-Methylenebisacrylamide | Induce cross-linking between lengthy polymer chains to type a three-dimensional community. |
| Tris-HCl Buffer | Maintain a secure pH. |
| APS | Facilitate cross-linking and supply free radicals that promote the polymerization of acrylamide and N,N-Methylenebisacrylamide. |
| TEMED | Catalyze the formation of free radicals and speed up polymerization. |
4.2 Electrophoresis Buffer Preparation
Conventional SDS-PAGE is a sturdy software for separating proteins with molecular weights (MWs) starting from 30 kDa to 250 kDa. Choose acceptable separation gel focus in line with the molecular weights of the proteins (Table 5).
Table 7. The parts of electrophoresis buffer (1L)
| Reagents | Mass (g) |
|---|---|
| Glycine | 14.4 |
| Tris | 3 |
| SDS | 1 |
| Add ddH2O and blend totally, pH 8.3. |
Use fixed energy within the Glycine-Tris-gel electrophoresis system, run 60-80 V for the concentrated gel, and 100-120 V for the separation gel. The decrease the voltage, the slower it runs, and a greater separation may be obtained.
However, the traditional Glycine-Tris-gel system doesn’t lead to decision for the separation of small molecular proteins with low molecular weights (<30 kDa). Tricine has higher electron mobility and dissociation fixed than Glycine, which gives small molecule proteins higher focus impact within the concentrated gel and better decision within the separation gel.
Table 8. Recommended electrophoresis buffers for small molecule proteins
| Reagent | Mass(g) | Comments | |
|---|---|---|---|
| Anode buffer(1L buffer system) | Tris | 24.228 | Dissolve Tris in ddH2O, combine totally and modify pH to eight.9. |
| Cathode buffer(1L buffer system) | Tricine | 17.92 | Dissolve reagent in ddH2O, combine totally. |
| Tris | 12.114 | ||
| SDS | 1 |
We suggest working Tricine-Tris-gel at fixed voltage (60-100 V).
4.3 Protein Marker and Loading Control
It’s vital to make use of acceptable controls based mostly on experimental requirement.
Indicator of molecular weight: Protein Marker that covers an acceptable vary of molecular weights can point out the molecular weight of the protein and to some extent replicate the electrophoresis impact and the membrane switch effectivity. Based on totally different options, the generally used Markers may be roughly divided into three varieties: unstained marker, pre-stained marker, and publicity marker.
Table 9. Comparison of various protein markers
| Protein Marker | Advantage | Disadvantage |
|---|---|---|
| Unstained marker | Unstained marker is essentially the most correct protein marker, it doesn’t carry dye molecules or labeling molecules and might precisely decide protein dimension. | Unstained markers usually are not seen throughout electrophoresis, electrophoresis and switch course of can’t be monitored in actual time. It have to be stained to be seen. |
| Prestained marker | Pre-stained Marker is a mix of proteins covalently coupled to dye, it may be immediately noticed by the bare eye throughout the experiment, and serves as a reference in electrophoresis and switch course of. | Due to the dye coupling, migration effectivity of the protein molecules after dyeing in electrophoresis is modified, leading to a shift of the indicated molecular weight, which may trigger inaccurate protein sizing. The ultimate consequence requires a comparability between the Marker and goal bands, throughout which human errors could happen. |
| Exposure marker | The goal bands and the Marker bands may be concurrently uncovered to scale back errors and the mobility of proteins. | High price. |
This explains why the noticed band dimension is totally different from the expected dimension in western blot. To decrease the influence of molecular weight shift brought on by prestained marker, examine it with unstained marker to determine the distinction in molecular weight for correct molecular weight sizing of the goal band (The left determine beneath signifies the distinction between prestained marker and unstained marker). In addition, the protein marker varies extensively from producer to producer, and prospects must be cautious when selecting protein marker. (The proper determine beneath signifies the distinction between prestained markers from totally different producers). When figuring out proteins in constructive western blot consequence, the molecular weight shift brought on by prestained marker must be considered.
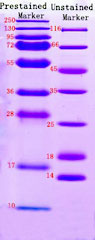
Prestained maker and unstained marker
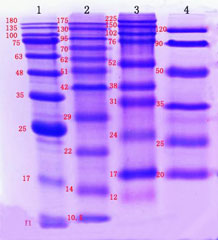
Prestained markers from totally different producers
Positive Control: tissues or lysate from cell traces that specific the protein of curiosity.
Loading Control: stage of the proteins encoded by the housekeeping genes are comparatively fixed in varied tissues and cells, such protein can function loading management throughout the quantification of the protein of curiosity to make sure the identical quantity of protein is loaded into every lane. In addition, the loading management protein can be utilized to evaluate whether or not the experiment runs efficiently.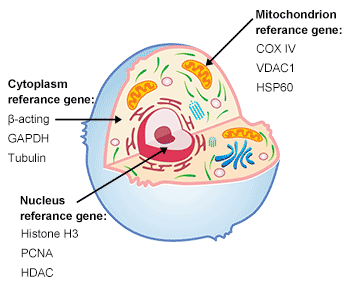
Fig. 3. The construction of cell
Common inside reference gene (Housekeeping gene): It is essential to decide on the correct loading management protein in line with the aim of the experiment when totally different proteins are studied.
The following components must be thought of when selecting loading management protein:
Table 10. The genes in several location of cell
| Nuclear | Nuclear membrane | Cytoplasm | Cellular membrane | Mitochondrion | Membrane | |
|---|---|---|---|---|---|---|
| Animal Tissue/Cell | Histone H3 (17 kDa), PCNA (29 kDa) | Lamin B (66 kDa) | β-actin (43 kDa), GAPDH (36 kDa), Tubulin (5 kDa) | Na+/Ok+-ATPase (120 kDa) | CoX IV(17 kDa), VDAC1 (30 kDa) | ATP1A1 (113 kDa) |
a. The expression stage of some housekeeping genes could range in response to sure experimental situations, similar to exterior stimulation or drug therapy. When selecting a loading management protein, it is vital to seek the advice of related literature and validate that its expression is fixed throughout samples and never affected by sure experimental situations.
b. The molecular weight of the protein of curiosity must be totally different types that of the loading management protein to allow distinct detection and differentiation. If the loading management protein has an identical molecular weight to the protein of curiosity, visualize the band of protein of curiosity first, then wash the antibodies away with a main/secondary antibody removing answer, adopted by the incubation with loading management antibody and the visualization of loading management protein.
4.4 Running the Gel
a. After flash spinning the samples to take away impurities, 10-30 μg of whole protein is advisable to load into the wells. The advisable amount of purified protein is 10-100 ng. Loading portions must be adjusted in line with the consequence.
b. Molecular weight markers must be included in a lane to point the protein of curiosity.
c. Run with fixed voltage (voltage set at 120 V or decrease). To receive a greater experimental consequence, 80 V is advisable when the entire protein migrates in focus gel. While the proteins migrate into separation gel, voltage must be boosted up.
d. Usual working time is about 1.2 h. However, the protein which weighs decrease than 20 kDa ought to shorten the working time in line with the fascinating protein. While the protein is larger than 100 kDa, working time must be prolonged to get a greater separation of protein.
5. Transfer
5.1 Transfer Methods
To switch the separated proteins from the gel to the strong part medium, moist switch and semi-dry switch strategies are generally used. The two switch strategies are an identical in precept, besides that the mechanical units for making use of the electrical area and the approaches to immobilize gels and membranes are totally different, semi-dry switch employs multi-layer filter paper infiltrated with buffer answer.
Table 11. Comparison between forms of transferring strategies
| Methods | Advantages | Disadvantages |
|---|---|---|
| Tank switch | Membrane switch result’s good, the temperature is controllable whereas transferring. | It takes longer time, and wish a big amount of switch buffer. |
| Semi-dry switch | Membrane switch effectiveness is excessive inside a short while, and it prices much less switch buffer. | The temperature can’t be managed whereas membrane transferring, thus the excessive temperature causes excessive background finally. |
While membrane transferring, the warmth generates quickly within the meeting inside a short while underneath excessive present. So it’s a necessity to take measures whereas membrane transferring to maintain a low temperature situation. While tank transferring, the meeting may be ice bathed for warmth dissipation, whereas semi-dry transferring, it is unsuitable to have a very long time electrical flip, in order that we suggest tank switch for prime molecular weight protein (Above 100 kDa).
As for low molecular weight and center molecular weight protein, the effectiveness of semi-dry switch and tank switch practically similar.Notes:
We suggest semi-dry switch for small piece of gel with excessive abundance.
We suggest tank switch for large piece of gel with low abundance.
5.2 Selection for Membranes
The nitrocellulose (NC) and polyvinylidene difluoride (PVDF) are essentially the most generally used membranes.
Table 12. Comparison between NC and PVDF membrane
| NC membrane | PVDF membrane | |
|---|---|---|
| Protein binding capability | 80-100 μg/cm2 | 100-300 μg/cm2 |
| Binding Strength | Low | High |
| Physical traits | Fragile | Durable and resilient |
| Whether activation is required | No want activation | Alcohol activation |
The binding means of NC membrane is especially associated to its purity, the upper purity is, the stronger protein binding means will likely be. However, the excessive purity NC membrane is fragile and straightforward to interrupt. Compared with NC membrane, PVDF membrane not solely has stronger protein binding means, but in addition owns higher chemical resistance.
Please notice that earlier than utilizing PVDF membrane, it must be soaked in methanol (>15 sec) to activate the positively charged teams on the membrane and equilibrated within the membrane buffer for a time period. Besides, the PVDF membrane and NC membrane have totally different pore sizes.
For small molecular protein (<20 kDa), we advise to undertake 0.22 μm of membrane pore dimension to keep away from exceed flip round.
For most of scenario, 0.45 μm of membrane pore dimension is recommended.
5.3 Protein Separation and Membrane Transfer Condition Optimization
Regarding to membrane switch for small molecular proteins, we’re succesful to optimize based mostly on the beneath features:
a. Increase the focus of cross-linking agent, and undertake 15% acrylamide gel to have electrophoresis. However, aimed on the proteins that beneath 15 kDa, the decision of 15% acrylamide gel is low, so we advise so as to add 10% of interlayer gel between the stacking gel and separating gel to extend the decision of small molecular proteins.
b. Replace Tris-Glycine buffer system with Tris-Tricine electrophoresis system to realize higher focus and separation results. Please notice that the voltage must be respectable whereas utilizing Tris-Tricine, 60 V-80 V will likely be good.
c. Choose 0.22 μm of pore dimension membrane.
d. Shorten the membrane switch time.
Regarding to membrane switch for giant molecular proteins, we’re succesful to optimize based mostly on the beneath features:
a. Decline the focus of cross-linking agent, course of the electrophoresis with 8%-5% acrylamide gel. Please notice that the decrease gel focus is, the extra fragile the membrane will likely be, please take cautious whereas operation.
b. While membrane transferring, please decently flip up the present, delay the membrane switch time and keep away from warmth generated. Tank switch at 4℃ temperature for in a single day is advisable.
c. Declining the methanol within the switch buffer facilitates to the separation of SDS molecule from the protein. Methanol with excessive focus fixates the protein and it is not good for giant molecular protein, it is suggested to say no methanol focus to 10%.
5.4 Transfer Efficiency Monitoring
a. The switch results of prestained marker displays protein switch effectivity.
b. Stain the gel with Ponceau, to evaluate whether or not the switch is profitable from stained bands. The course of is reversible, but it surely’s not appropriate for Nylon.
Fermentation of Ponceau staining answer: combine the 5% (V/V) acetic acid, 0.1% (W/V) Ponceau, ddH2O nicely, and saved at 4℃.
Ponceau staining course of:
- Soak the transferred PVDF or NC membrane to the Ponceau staining answer and oscillate for 5-10 min.
- Take out the imprinted membrane, wash Three instances × 5 min with PBS.
- Observe the stained crimson bands and make file to the switch outcomes.
- Wash Three instances×5min with PBS once more to take away the mixed Ponceau with a purpose to have an extra WB evaluation.
c. Stain the gel with Coomassie Blue Staining Solution, the method is irreversible, however the sensitivity of Coomassie Blue Staining Solution is greater than the sensitivity of Ponceau.
Fermentation for Coomassie Blue Staining Solution: Add 10% (V/V) glacial acetic acid, 45% (V/V) methanol, 0.25% (W/V), ddH2O and blend nicely.
Fermentation for Commassie Blue staining destaining answer: Add 25% (V/V) methanol, 8% (V/V) glacial acetic acid, ddH2O and blend nicely.
Commassie staining course of:
- Soak the transferred PVDF and NC membrane to the Coomassie Blue Staining Solution, use a shaker to shake slowly at room temperature for 1 h (Advice to make some changes in line with the gel dimension, thickness, and temperature) till the gel turns blue.
- Pour out the staining answer and soak the gel to the destaining answer, shake slowly with a shaker at room temperature for Four h till the blue background is destained and the protein bands are seen.
5.5 Preparation of Transfer Buffer
The fermentation of 1L switch buffer as beneath:
Table 13. The parts of 1L switch buffer
| Reagents | Mass (g) |
|---|---|
| Glycine | 11.26 |
| Tris | 2.43 |
| Methanol | 200 mL |
| *Add ddH2O and blend nicely to dissolve sufficiently |
The switch buffer must be saved away from mild, it may be repeated utilizing.
However, as a result of methanol is risky, it’s a necessity to vary to the recent switch buffer in time.
5.6 Transfer
a.Be cautious to separate the separation gel.
b. Make the “sandwich” by filter paper-gel- filter paper, preserve the “sandwich” hydrated and keep away from bubble between it. Prepare the “sandwich” as follows: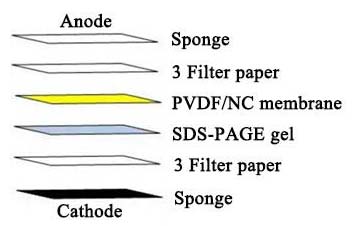
c. Assemble the switch machine in line with producer’s guide. Transfer proteins to nitrocellulose or PVDF membrane. 0.22 μm membrane is advisable for the protein which molecular weight decrease than 20 kDa.
d. There are often two units that you may select within the mild of precise situations, moist switch or semi-dry-transfer. Both moist switch and semi-dry-transfer can work nicely for standard dimension protein (20-100 kDa), whereas moist switch is extra appropriate for prime molecular weight proteins given that semi-dry-transfer can yield greater background staining.
e. Rinse the blot in PBS for roughly 5 min.
6. Blocking
Block the membrane utilizing blocking buffer for 1h at room temperature.
Blocking buffer choice
The binding floor is uneven, there are many small holes. While the protein is being transferred to binding floor, it absorbs to the binding floor via mutual impact. However, not all the websites are being absorbed to proteins, in order that the blocking buffer is required to soak up to the remainder of binding websites, with a purpose to stop the antibody molecular from absorbing to the membrane immediately and trigger the faux constructive or excessive background consequence.
The precept for choosing a blocking buffer must be:
a. The blocking buffer is succesful to dam all the uncombined websites on the blocking membrane.
b. The blocking buffer would not intervene the mix of goal proteins. It would not mix with the epitopes of goal proteins, or cross-react with different reagents.
Here are particular particulars of blocking buffers that are generally used:
Table 14. Comparison of assorted blocking buffer
| Blocking buffer | Advantages | Disadvantages |
|---|---|---|
| 5% Skim milk powder | The element is advanced, it accommodates many proteins with totally different molecular weight, which may sufficiently blocking. | It would not appropriate for biotin-avidin and alkaline phosphatase detection techniques (on account of small quantities of biotin and alkaline phosphatase residues existed in skimmed milk powder) |
| 1% Casein | It has unfavorable cost in Neutral and alkaline situation and has interplay with positively charged membranes. | The solubility is comparatively unhealthy, and it is not good for blocking. |
| 5% BSA | The element is straightforward, it is appropriate for many of conditions. | When the immunogen is coupled with BSA, it could cross-react with the residual BSA within the antibody on account of its sure immunogenicity, leading to a sure background. |
| Serum | Not solely blocks non-specific binding, but in addition the antibodies in serum block FC receptors that presumably exist within the pattern to keep away from the first and secondary antibody react towards the FC receptor. | The price is comparatively greater. |
| Non-protein compound | Gelatin, Tween-20, and so on. can cut back the hydrophobic interplay between proteins, elute non-specific adsorption, and enhance the particular recognition means of antibodies. |
A comparative experiment for various blocking buffers is carried out as beneath: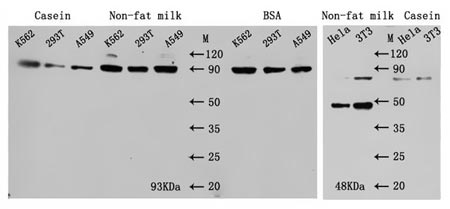
Fig. 4. Comparison of various blocking bufferNotes:
It is vital to notice that for phosphorylated proteins, skim milk powder and casein usually are not advisable as blocking buffer, and we do not counsel to interchange PBST with TBST both. The choice for blocking buffers ought to have some changes in line with the totally different outcomes. For most of antibodies, undertake skim milk powder as blocking buffer reaches good blocking impact, nonetheless, undertake BSA as blocking buffer for among the antibodies will likely be good for decreasing the background. Furthermore, often the blocking situation is underneath the room temperature for 1 h.
7. Primary Antibody Incubation
Obey the product protocol and dilute the first antibody, often the parts of main antibody dilution buffer and blocking buffer are similar. Besides, we advise to decide on validated main antibodies, and the first antibody must be incubated at 4℃ for in a single day, in order that the antigen and antibody might mix sufficiently.
We advise to have a gradient preliminary experiment to find out one of the best dilution ratio of main antibodies, like Dot blot technique.
a. Upload the samples with totally different loading quantity sequentially on the NC membrane and air dried naturally.
b. Block the membrane after absorbing the pattern fully.
c. Cut the membrane in line with the loading gradient.
d. Incubate the first and secondary antibodies with totally different focus gradient respectively.
e. At final, undertake ECL luminescent substrate incubation and improvement to preliminary decide the dilution ratio vary based mostly on the event consequence.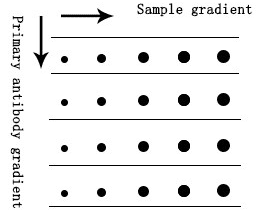
Fig.5 Dot Blot
8. Secondary Antibody Incubation
8.1 Experimental Operation
a. Before the secondary antibody incubation, wash the membrane 3×10 min with PBST/TBST to remove the uncombined main antibodies.
b. Properly dilute the secondary antibody, incubate at room temperature for 1 h.
c. After the secondary antibody incubation completed, wash the membrane 3×10 min with PBST/TBST to remove the uncombined secondary antibodies.
8.2 Selection For Secondary Antibody
- Species useful resourceWe don’t recommendation to pick secondary antibodies with rabbit, rat or mouse species, as a result of their homology is way comparable with human species and straightforward to have cross-reactivity, then induced excessive background finally. Secondary antibodies in goat or donkey species are generally used, please notice that species useful resource of the chosen secondary antibodies have to be totally different with the chosen main antibody, the secondary antibody species useful resource choice relies on the species of main antibody.Besides, it is also crucial to concentrate on the subtype should you have been utilizing a monoclonal main antibody, and the chosen secondary antibody which towards the subtype of main antibody.
- Purification techniqueThe primarily purification strategies are Protein G/A purification and Antigen affinity purification.Protein G/A purification technique combines all the antibody IgG molecular in serum, there isn’t a distinguish with antigen specificity.Affinity purification is a technique of eluting by binding to a ligand or receptor particularly acknowledged by an antibody. And a particular antibody element in serum may be purified through the strategy. So secondary antibodies with antigen affinity technique will decline the unspecific binding and enhance the specificity of detected proteins.
- Suitable conjugationsIn WB validation, essentially the most generally used conjugation for secondary antibody is HRP conjugate, similar to Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP).Commonly used as substrate, HRP has the attribute of excessive specificity, secure, speedy and economical. Although AP is extra delicate, the background is often excessive, and the endogenous phosphatase which will exist within the experimental pattern and intervene consequence.Furthermore, whereas utilizing AP conjugation for secondary antibody, please be fastidiously to decide on the blocking buffer with a purpose to keep away from phosphatase interference.
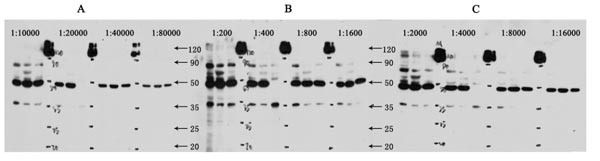
Fig. 6. The dilution ranges are in line with directions of various manufactures
9. Image Development
Chemiluminescence improvement
Luminol, which is without doubt one of the most classical HRP chemiluminescence substance, can generate enzyme catalysis response with Horseradish peroxidase within the presence of H2O2, it has excessive sensitivity and good imaging attribute, which may develop on the movie.
Substrate improvement
There are many sorts of HRP chromophoric substrates, essentially the most generally used is DAB. It develops by reacting with HRP to type an insoluble brown precipitate, and owns excessive sensitivity as nicely.
However, it must be used on spot and it’s carcinogenic, so please be careful whereas working.
Fluorescent improvement
By utilizing an acceptable fluorescent secondary antibody, fluorescence secondary antibody improvement may be achieved, which makes up the quantitative defects of chemiluminescence and substrate improvement.
10. Frequently Asked Questions
10.1 High Background
| Possible Cause | Solution |
|---|---|
| Insufficient blocking or inappropriate blocking buffer. | Optimize the blocking impact and choose an accurate blocking buffer. |
| The focus of incubated antibody is simply too excessive, incubation time is simply too lengthy or the temperature is simply too excessive. | Adjust the antibody incubation focus and incubation time. |
| Inadequate washing. | Increase the numbers to scrub, and prolong the washing time. |
| Secondary antibody non-specifically binding. | Set a management for the secondary antibody and choose a acceptable secondary antibody. |
| Membrane is dry. | Keep the membrane moist whereas working. |
| Membrane is contaminated. | Keep the membrane clear whereas working, don’t press by hand. |
| Excess chemiluminescent substrate residue. | Drain the surplus chemiluminescent liquid then develop. |
| Film publicity time is simply too lengthy. | Check a number of instances to verify optimum publicity time. |
10.2 No Target Bands
| Possible Cause | Solution |
|---|---|
| The goal protein in pattern expresses with low abundance, or it has no expression in any respect. | Double affirm the feasibility of detected pattern, enrich the abundance of the goal protein earlier than detecting. |
| Protein degradation throughout extraction. | During protein extraction, preserve protein at low temperature and add protease inhibitor. |
| The incorrect storage situation for protein samples and trigger the protein degradation. | The protein samples are prompt to retailer after thermal denaturation with SDS. Valuable samples are prompt to retailer at -80℃. |
| Protein switch effectivity is low. | Adopt Ponceau to verify if the switch system is regular. |
| Antibody incubation focus is simply too low, or incubation time is simply too quick. | Optimize the quantity of antibody incubation, main antibody is advisable to incubate in a single day at 4℃. |
| Primary and secondary antibody usually are not appropriate. | Select right main antibody and secondary antibody. |
| The antibody is inactivated. | Store the antibodies correctly. |
| The movie improvement answer could have expired. | Use recent movie improvement options, and stop from mild. |
10.3 The Observed Band Size Don’t Match with the Predicted Band Size
| Possible Cause | Solution |
|---|---|
| Marker’s indicated dimension has deviation. | Choose an acceptable prestained marker and observe the distinction with the unprestained marker. |
| The affect of electrophoresis system. | To keep away from the instability components of the sting wells, load the pattern to the center wells, and add the identical quantity of pattern loading buffer into the sting wells electrophoresis to stability system. |
| Post-translational modifications. | Look via the literature to double affirm if the protein is phosphorylated or glycosylated, and so forth. |
| Post-translational shear and isomers. | Look via the literature to see if the protein has a number of splicing energetic types. |
| Formation of protein polymer. | Keep protein monomer standing by utilizing recent DTT or β-mercaptoethanol throughout pattern preparation. |
| Relatively change of protein expenses. | The amino acid composition of the protein is totally different, and the cost of some proteins shouldn’t be fully being coated by negatively charged SDS, ensuing within the protein migration fee shouldn’t be proportional to the protein dimension. |
10.4 Other Issues
| Possible Cause | Solution |
|---|---|
| Reflection or yellow bands on membrane is being discovered. | The focus of main or secondary antibody is simply too excessive, or the pattern loading amount is an excessive amount of, induced the enzymes is being consumed instantaneously. |
| White clean level. | There are bubbles stays within the transmembrane for sandwich, or the antibody shouldn’t be evenly incubated, so it must be oscillated incubate. |
| Black dots on the background. | The granule of blocking buffer residues, please stir and dissolve the granule sufficiently earlier than utilizing. |
| Smiling bands. | The migration was too quick, lower the voltage whereas working the gel. The gel solidifies erratically, it must be ready appropriately. |
| Frowning bands. | When electrophoresis, the substrate could mixture plenty of bubbles, it is easy to steer voltage imbalance and trigger frown bands finally. |
| Smeared bands. | The pattern accommodates insolubles, we advise to centrifuge or optimize the protein extraction. Sample overload, please load much less protein into every lane. The focus of the gel shouldn’t be appropriate, the protein decision is low. We advise to regulate the ratio of cross-linking agent. The electrophoresis buffer could also be repeat utilizing, please change to recent electrophoresis buffer. |
| Distorted bands. | The floor of gel is uneven and or the gel is ready erratically. The focus of salt ion in pattern is simply too excessive, it interferes the electrophoresis. The voltage is simply too excessive, in order that it results in quick migration. |