

BACKGROUNDHigh density genotyping knowledge are indispensable for genomic analyses of advanced traits in animal and crop species.
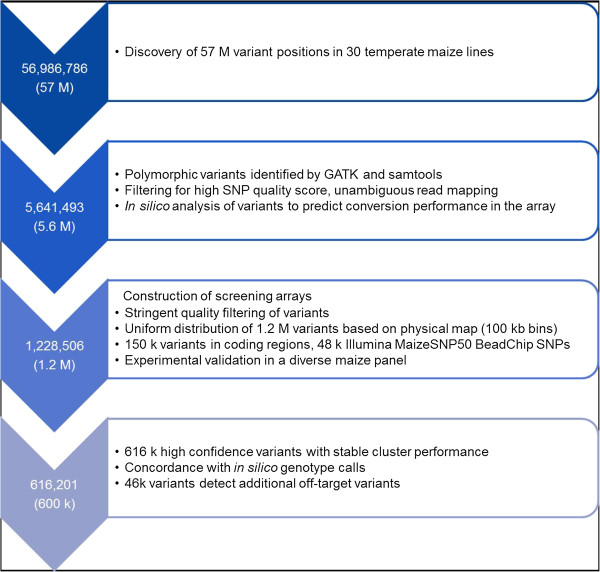
Maize is one of the most vital crop vegetation worldwide, nevertheless a high density SNP genotyping array for analysis of its giant and extremely dynamic genome was not out there to date.RESULTSWe developed a high density maize SNP array composed of 616,201 variants (SNPs and small indels).
Initially, 57 M variants had been found by sequencing 30 consultant temperate maize strains and then stringently filtered for sequence high quality scores and predicted conversion efficiency on the array ensuing in the choice of 1.2 M polymorphic variants assayed on two screening arrays.
To establish high-confidence variants, 285 DNA samples from a broad genetic range panel of worldwide maize strains together with the samples used for sequencing, vital founder strains for European maize breeding, hybrids, and proprietary samples with European, US, semi-tropical, and tropical origin had been used for experimental validation.
We chosen 616 k variants in accordance with their efficiency throughout validation, help of genotype calls by way of sequencing knowledge, and bodily distribution for additional analysis and for the design of the commercially out there Affymetrix® Axiom® Maize Genotyping Array.
This array consists of 609,442 SNPs and 6,759 indels. Among these are 116,224 variants in coding areas and 45,655 SNPs of the Illumina® MaizeSNP50 BeadChip for examine comparability.
In a subset of 45,974 variants, other than the goal SNP extra off-target variants are detected, which present solely a minor bias in the direction of intermediate allele frequencies.
We carried out principal coordinate and admixture analyses to find out the potential of the array to detect and resolve inhabitants construction and investigated the extent of LD inside a worldwide validation panel.CONCLUSIONSThe high density Affymetrix® Axiom® Maize Genotyping Array is optimized for European and American temperate maize and was developed primarily based on a various pattern panel by making use of stringent high quality filter standards to make sure its suitability for a broad vary of functions. With 600 k variants it’s the largest presently publically out there genotyping array in crop species.
Variant affiliation instruments for high quality management and analysis of large-scale sequence and genotyping array knowledge.
Currently there’s nice curiosity in detecting associations between advanced traits and uncommon variants.
In this report, we describe Variant Association Tools (VAT) and the VAT pipeline, which implements finest practices for rare-variant affiliation research. Highlights of VAT embody variant-site and call-level high quality management (QC), abstract statistics, phenotype- and genotype-based pattern choice, variant annotation, choice of variants for affiliation analysis, and a set of rare-variant affiliation strategies for analyzing qualitative and quantitative traits.
The affiliation testing framework for VAT is regression primarily based, which readily permits for versatile development of affiliation fashions with a number of covariates and weighting themes primarily based on allele frequencies or predicted performance. Additionally, pathway analyses, conditional analyses, and analyses of gene-gene and gene-environment interactions may be carried out.
VAT is succesful of quickly scanning by way of knowledge through the use of multi-process computation, adaptive permutation, and concurrently conducting affiliation analysis by way of a number of strategies.
Results can be found in textual content or graphic file codecs and moreover may be output to relational databases for additional annotation and filtering. An interface to R language additionally facilitates consumer implementation of novel affiliation strategies.
The VAT’s knowledge QC and association-analysis pipeline may be utilized to sequence, imputed, and genotyping array, e.g., “exome chip,” knowledge, offering a dependable and reproducible computational atmosphere in which to research small- to large-scale research with knowledge from the newest genotyping and sequencing applied sciences.
Application of the VAT pipeline is demonstrated by way of analysis of knowledge from the 1000 Genomes venture.